Rétinoblastome
Tout savoir sur le rétinoblastome
Le rétinoblastome est une tumeur maligne primitive de la rétine. Il s’agit du cancer intraoculaire le plus fréquent chez l’enfant.
L’apparition d’un rétinoblastome est liée à la mutation du gène RB1, un gène suppresseur de tumeur. Cette mutation peut apparaitre au hasard dans une cellule précurseur de la rétine mais peut également être présente à l’état constitutionnel (dès la naissance) dans l’ensemble ou une partie des cellules de l’enfant. Le rétinoblastome survient ainsi pour la moitié des cas dans un contexte de prédisposition génétique.
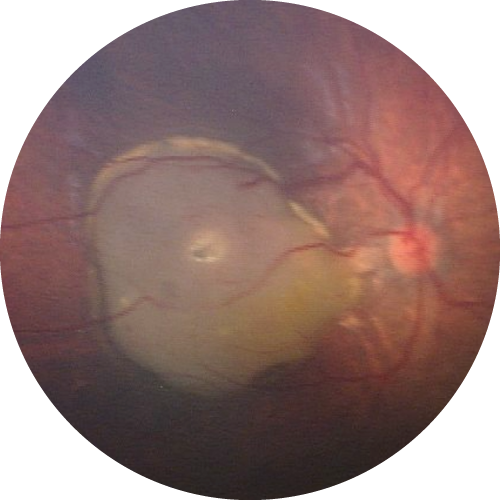
En dehors des enfants surveillés dès la naissance dans le cadre d’un programme de surveillance personnalisé, le diagnostic reste trop souvent tardif, par l’apparition d’une pupille blanche (leucocorie) ou d’un strabisme, devant faire réaliser un examen ophtalmologique avec examen du fond d’œil dilaté dans les meilleurs délais. Le diagnostic est ensuite confirmé en centre de référence à l’Institut Curie à Paris. En cas de prédisposition génétique, une atteinte des deux yeux est possible.
La précocité du diagnostic conditionne la mise en œuvre de traitements conservant au mieux les yeux et préservant la meilleure vision possible selon l’atteinte initiale. Dans certains cas, le traitement peut nécessiter l’ablation de l’œil (énucléation).
La surveillance ophtalmologique sera précisée par l’onco-ophtalmologue référent, suivant les traitements entrepris et la présence d’une prédisposition génétique. Une étude génétique chez les apparentés indemnes de la maladie mais potentiellement porteurs de la mutation est habituellement proposée, dans le but de mettre en place une surveillance ophtalmologique spécifique.